K Domino, MB Johansen, K Daasbjerg, T Skrydstrup
https://doi.org/10.1021/acs.organomet.9b00849
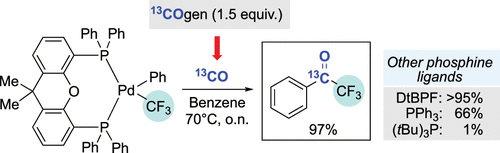
We have performed a series of stoichiometric studies in order to identify viable steps for a hypothetical catalytic cycle for the palladium-mediated carbonylative coupling of an aryl bromide with TMSCF3. Our work revealed that benzoyl Pd(II) complexes bearing Xantphos or tBu3P as the phosphine ligands, which are generated from the corresponding PdII(Ph)Br complexes exposed to stoichiometric 13CO from 13COgen, were unable to undergo transmetalation and reductive elimination to trifluoroacetophenone. Instead, in the presence of base and additional CO, these organometallic complexes readily underwent reductive elimination to the acid fluoride. Attempts to determine whether the acid fluoride could represent an intermediate for acetophenone production were unrewarding. Only in the presence of a boronic ester did we observe some formation of the desired product, although the efficiency of transformation was still low. Finally, we investigated the reactivity of four phosphine-ligated PdII(Ph)CF3 complexes (Xantphos, DtBPF, tBu3P, and triphenylphosphine) with carbon monoxide. With the exception of the tBu3P-ligated complex, all other metal complexes led to the facile formation of trifluoroacetophenone. We also determined in the case of triphenylphosphine that CO insertion occurred into the Pd–Ar bond, as trapping of this complex with n-hexylamine led to the formation of n-hexylbenzamide.